
New molecules and crystal structure models.
Our catalogue comprises over 2000 structures that we have built previously but as the ICSD alone contains over 90,000 inorganic structures and there are millions of organic compounds, it is inevitable that people will often want a structure or molecule that we haven't built before.
![a large crystal structure model of the mineral Bechererite, (Zn,Cu)6Zn2(OH)13[(S,Si)(O,OH)4]2](../../images/bechlikeb3.jpg)
Bechererite (Zn,Cu)6Zn2(OH)13[(S,Si)(O,OH)4]2 - a mineral so unusual most people have never heard of it, so there was no chance it was going to be in anyone's catalogue...
That's no problem to us - even if we haven't made the structure before, we'll be able to produce a model of it. If it exists in nature, there is rarely any reason why a model of it cannot be built. In all cases, we'll discuss the model with you to confirm what you want. If you are interested in a model of a new material, it may be worth considering the following aspects, as these are things we will discuss with you:
Model size
For most crystal structures, we make a model that is around 1.3-1.5x each unit cell length in size - that normally generates a complete coordination sphere around each atom type so that you can fully appreciate the different environments that each atom occupies. Some structures do not lend themselves to this size, though - in particular, structures that are elongated along one axis, with very short axes along the other two may result in a model that is very fragile - in some cases, there may be only one or two bonds holding the two halves of the model together. In these instances, we tend to advise inclusion of two or three unit cell lengths along the shorter axes. Having said that, we can make the model as large or as small as you wish - larger models look amazing.
Structure of the molecule v. its structure in the crystal
This can be a common source of misunderstanding - people very often send us files of organic molecules or metallo-organic molecules within crystals as cif files (for example), simply wanting a model of a single molecule. No problem with that, but then we ask whether they really want, for example, the structure they actually provided, with squashed and mis-shapen phenyl rings, or whether they want an idealised phenyl ring with trigonally coordinated carbon atoms.
The issue is that most people expect ideal the tetrahedral and trigonal planar geometries of VSEPR theory in their molecules and very often don't look that closely. But VSEPR only works in isolated molecules and, in crystals, they aren't isolated - near neighbours influence the shape and position of molecular groups in the compound. Many people actually prefer idealised side groups, while for others it matters that they are accurately represented - it's worth considering which you prefer.
Partial and mixed occupancy
In many materials, partial occupancy of some sites, or mixed occupancy by two or more atom types, will be a feature. Normally, this will result in a random distribution of atoms in that site - paired with either vacancies or atoms of another type. The latter type is commonly seen with Al/Si distributions in silicate minerals. Normally, we choose which atoms to use (or whether to use one at all) literally picking balls at random from a pot - this can cause confusion in some customers, because the resulting arrangement can, ironically, be perceived as non-random. So, for example, with atoms A and B on a site, each with partial occupancy of 0.5, it is perfectly possible to have three or four 'A' atoms on neighbouring sites - in fact, across a large number of such sites in a model, counter-intuitive though it is, it is to be expected! If you want us to avoid such true random arrangements, please let us know.
Disorder in crystals
This can be a nightmare, but we can deal with it. In many cases, the disorder is relatively simply thermal disorder with, for example, a metal atom switching between two thermodynamically stable sites with a low kinetic barrier between them. In these cases, we will provide some suggestions to the customer and, with their agreement, devise an arrangement that gives a credible structure whilst, if possible, retaining the formal symmetry of the structure.
Sometimes, the disorder is more extreme and confuses the structure to the point where it is almost impossible to see the real form. An example is Vaterite (a polymorph of calcium carbonate). This structure is, in some cif files, riddled with disorder. By working with the structure and with the customer, we arrived at a very respectable structure that was later vindicated by more recent crystallographic data.
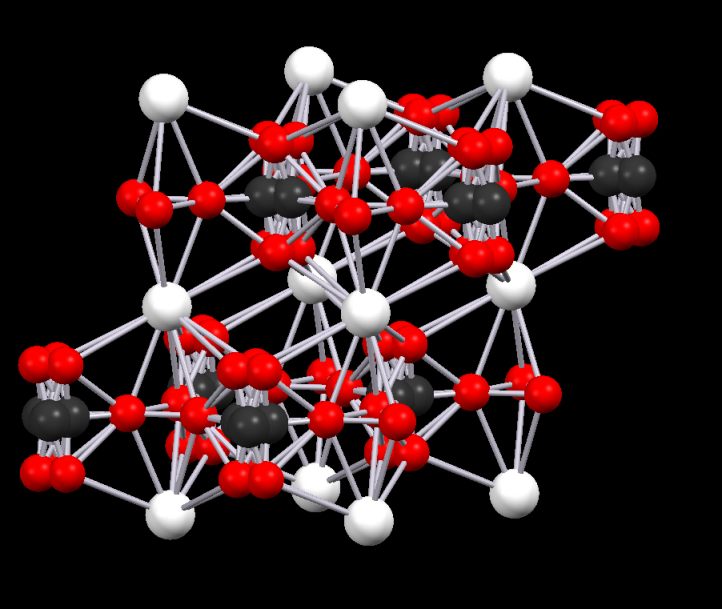

Disorder in the original cif file for vaterite (top) and the model created from it..
Company Registered in Scotland no. SC329683 VAT no 916067916